| OMIM: | 614962, 614963, 602025, 618406 |
| Diagnostik: | Sequenzierung und CNV: CACNA1C, CALM1, CALM2, CALM3, CASQ2, CAV3, GNB5, KCNE1, KCNE2, KCNE3, KCNH2, KCNJ2, KCNJ5, KCNQ1, RYR2, SCN4B, SCN5A, SNTA1, TECRL, TRDN |
| Material: | 2 ml EDTA-Blut |
| Analysezeit: | 6-8 Wochen |
| Formulare: |
Das angeborene familiäre LongQT-Syndrom (LQTS) ist eine seltene, genetisch bedingte Herzerkrankung und tritt mit einer Prävalenz von ca. 1:5.000-10.000 auf. Klinisch ist das LQTS durch polymorphe ventrikuläre Tachykardien charakterisiert, die über Kammerflimmern bis hin zum plötzlichen Herztod (SCD) führen können.
Beim LQTS kommt es zu funktionellen Störungen verschiedener Ionenkanäle der Zellmembran des Herzens, die zu einer verlängerten ventrikulären Repolarisation führen. Diese Repolarisationsstörung zeigt sich im Elektrokardiogramm (EKG) durch ein verlängertes korrigiertes QT-Intervall (QTc). Eine pathologische QTc-Zeit ist je nach Alter und Geschlecht unterschiedlich definiert und liegt bei Männern bei >470 ms, bei Frauen bei >480 ms und bei Kindern bei > 460 ms.
Das Krankheitsbild des angeborenen LQTS ist sehr heterogen und die Ausprägung der Symptome sind variabel. Bei bis zu 95% der LQTS-Patienten liegen pathogene Varianten in den Genen KCNQ1 (LQTS Typ 1; ~45%), KCNH2 (LQTS Typ 2; ~40%) und SCN5A (LQTS Typ 3; ~10%) vor. In seltenen Fällen lassen sich auch pathogene Varianten in weiteren Genen nachweisen.
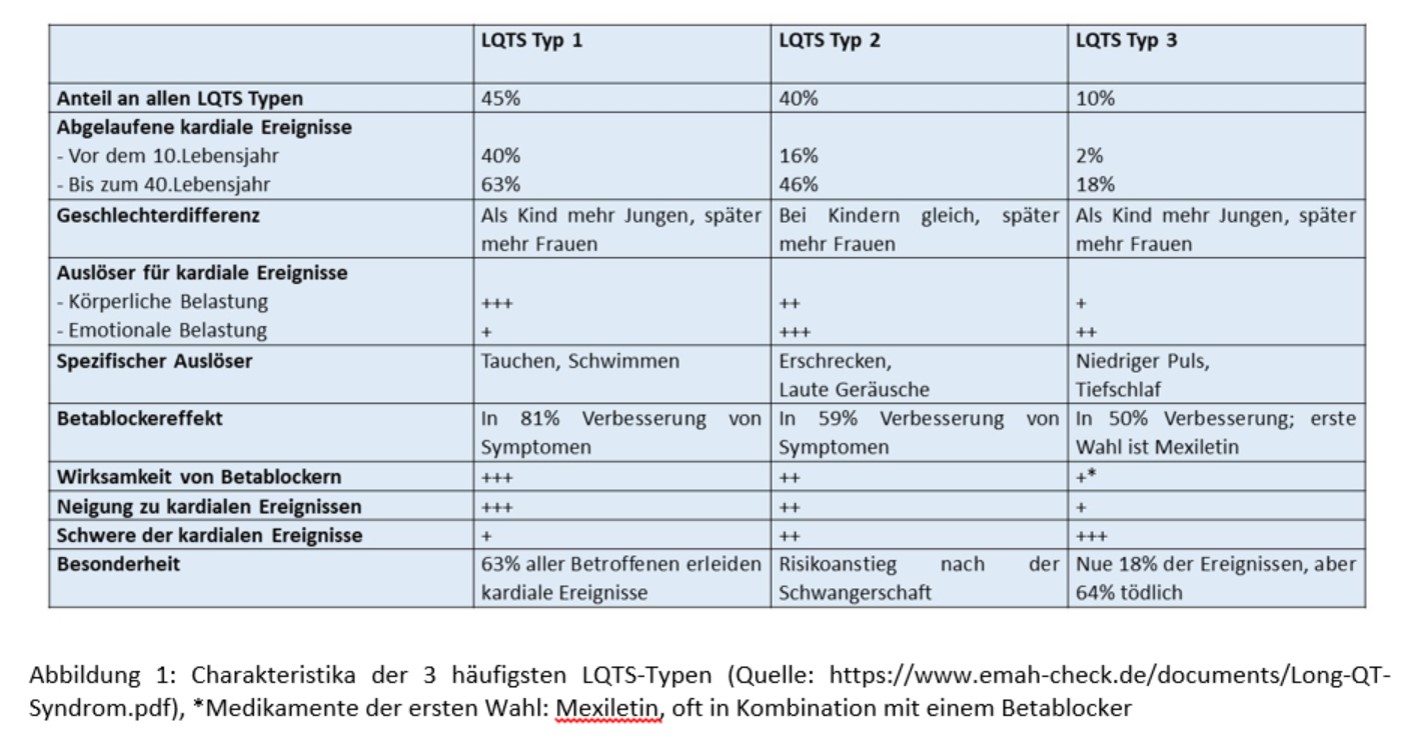
Diese 3 LQTS-Typen sind neben der verlängerten QTc-Zeit durch eine spezifische T-Wellen-Morphologie gekennzeichnet. Beim Typ 1 liegt eine breite T-Welle vor, Typ 2 ist durch eine doppelgipflige T-Welle charakterisiert und Typ 3 zeigt deutlich verspätete T-Wellen mit variabler Morphologie. Neben den zuvor beschriebenen LQTS Typen 1 bis 3 gibt es seltene Formen des LQTS, die u.a. durch Mutationen in den Genen KCNJ2, CACNA1, CAV3 oder SCN4B charakterisiert sind.
Die angeborenen familiären LQTS werden häufig autosomal dominant vererbt und als Romano-Ward-Syndrom bezeichnet. Deutlich seltener tritt das LQTS autosomal rezessiv auf. Bei diesen Patienten wird vom Jervell-Lange-Nielsen-Syndrom gesprochen, wobei aktuell zwei autosomal rezessive Formen bekannt sind und die Betroffenen zusätzlich eine angeborene Hörstörung aufweisen.
Die Ausprägung des Krankheitsbildes der LQTS-Patienten ist sehr variabel und reicht von keinen bis hin zu schweren Symptomen. Etwa 25% der Patienten mit einer nachgewiesenen LQTS-assoziierten Mutation sind stumme Mutationsträger und erleiden in ihrem Leben keine kardialen Ereignisse. Das Erkrankungsalter von LQTS-Patienten ist sehr unterschiedlich. Bei etwa der Hälfte aller Patienten liegt das Ersterkrankungsalter unter 12 Jahren. In seltenen Fällen können die ersten Symptome bereits im Säuglingsalter auftreten. Häufige Symptome der LQTS-Patienten sind u.a. plötzlich auftretende Unregelmäßigkeiten der Herzfrequenz und des Herzrhythmus, Schwindel oder Ohnmacht und Bewusstlosigkeit. Die schwerwiegendsten Symptome eines LQTS sind lebensbedrohliche Herzrhythmusstörungen bis hin zum akuten Herzversagen.
Neben der molekulargenetischen Analyse erfolgt die erste Diagnostik eines LQTS häufig mit Hilfe der Berechnung eines Scores, der auf den EKG-Veränderungen und der Familienanamnese des Patienten basiert. Je nach ermitteltem Punktescore wird eine Risikoeinschätzung für den Betroffenen in Form von gering (<1,5), möglich (1,5-3) und wahrscheinlich (>3,5) vorgenommen.
Ensprechend der Schwere der Symptomatik des Patienten erfolgt die Behandlung standardmäßig durch elektrische Kardioversion als Sofortmaßnahme, medikamentös insbesondere mit Betablockern, und/oder konservativ durch Vermeidung von Triggersituationen und einer Anpassung des individuellen Lebensstils.
Alternativ zu dem angeborenen LQTS kann es auch durch sekundäre Ursachen zu einer QTc-Verlängerung kommen wie z.B. medikamentös-induziert. Dafür sind insbesondere Veränderungen im CYP2D6, CYP2C9 und CYP2C19-Gen bekannt, die zu einem verzögerten Metabolismus der Medikamente und einem damit verbundenen verlängerten QTc führen können.