
Die Hochdurchsatz-Sequenzierung, auch als Next Generation Sequencing (NGS) genannt, ist eine parallele Sequenzierung von tausenden Nukleinsäure-Fragmenten, wobei Sequenzveränderungen wie z.B. Mutationen, Insertionen oder Deletionen detektiert werden. Dieses Verfahren ist mit der herkömmlichen Sanger-Sequenzierung nicht bzw. nur mit hohem Zeit- und Kostenaufwand möglich.
Beim NGS werden spezifische genetische Targetregionen (komplette Genome, Exome oder nur einzelne Gene wie z.B. BRCA1, BRCA2) von mehreren Patienten zeitgleich analysiert. Mit Hilfe von komplexen bioinformatischen Auswertungsalgorhythmen werden die DNA-Sequenzen den jeweiligen Patienten zugeordnet und hinsichtlich Genveränderungen im Vergleich zum Referenzgenom analysiert. Diese Genveränderungen können dann nach ihrer klinischen Relevanz mit Hilfe von Mutationsdatenbanken, in silico Tools und der aktuellen Literatur bewertet und in einem Befund zusammengetragen werden.
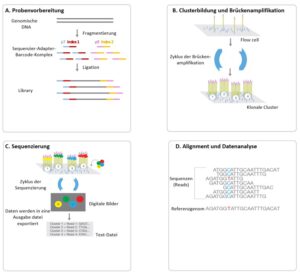
Abbildung 1: Schematische Darstellung zum Arbeitsablauf der Hochdurchsatz-Sequenzierung nach dem Sequencing By Synthesis-(SBS)-Verfahren der Firma Illumina
Unser Labor verfügt aktuell über zwei Benchtop-Sequenzer der Firma Illumina:
- MiSeq
- NextSeq
Zu Beginn der Probenvorbereitung wird die genomische DNA fragmentiert und anschließend werden universelle Sequenzier-Adapter und spezifische Barcodes (Index) für jede zu analysierende Probe an beide Enden der zu analysierenden genomischen DNA-Fragmente ligiert (Abbildung 1A). Barcodes sind einzigartige Sequenzen, die spezifisch an die zu analysierende Probe angefügt werden und es ermöglichen zeitgleich mehrere Proben zu analysieren. Die Sequenz-Adapter werden für die Bindung der zu analysierenden DNA-Fragmente an die Flowcell benötigt, wobei letztere einen Glasobjektträger mit immobilisierten Oligonukleotiden darstellt, worauf die Amplifikation und der Sequenzier-Prozess stattfindet. Anschließend werden die verschiedenen Proben-Fragmente inkl. Sequenzier-Adapter-Barcode-Komplexen zu einem Gesamtpool (sog. Library) zusammengeführt und auf die Flowcell geladen, wo die DNA-Fragmente komplementär an die immobilisierten Oligonukleotide der Flowcell binden (Abbilung 1B). Durch die Bindung der Sequenzier-Adapter an die Flowcell-Oligonukleotide entsteht eine DNA-Brücke, welche als Startpunkt für die DNA-Synthese (sog. Brückenamplifikation) dient. Anschließend folgen mehrere Zyklen Denaturierung, Renaturierung und Neusynthese, wodurch klonale Cluster mit einer hohen Dichte von identischen DNA-Fragmenten erzeugt werden. Nun beginnt der eigentliche Sequenzier-Prozess nach dem Verfahren der Sequenzierung bei Synthese (engl. Sequencing By Synthesis, SBS) (Abbildung 1C).
Beim MiSeq wird dazu die 4 Kanal-SBS-Technologie verwendet (siehe Abbildung 2) und 4 verschiedene mit Fluoreszenzfarbstoff und mit einer Blockierungsgruppe markierte dNTPs hinzugegeben, wobei diese entsprechend der Basenabfolge komplementär an die DNA-Fragmente durch die Polymerase eingebaut werden. Jeder Nukleotideinbau entspricht einem Zyklus und nach jedem Zyklus wird die Fluoreszenzintensität gemessen und dokumentiert. Die Anzahl der Zyklen entspricht der Sequenzierlänge (sog. Readlänge) der Sequenzierung, die von Library zu Library variiert und von der jeweiligen DNA-Fragmentlänge abhängig ist. Die Readlänge wird vor jeder Sequenzierung manuell in einem sog. SampleSheet angegeben. Die Sequenzierung und Analyse der DNA-Fragmente erfolgt zumeist von beiden Leserichtungen (3´- und 5´-Richtung), was als sog. Paired-End-Run bezeichnet wird.
Beim NextSeq erfolgt die Sequenzierung nach dem sogenannten 2 Kanal-SBS-Technologie, wobei ein Mix verschiedener mit Fluoreszenzfarbstoff und mit einer Blockierungsgruppe markierter dNTPs hinzugegeben wird und diese entsprechend der Basenabfolge komplementär an die DNA-Fragmente binden. Die Detektion erfolgt anschließend im Spektrum der grünen und roten Wellenlänge (siehe Abbildung 2). Die Detektion von 2 Farben bei 4 Nukleotiden sorgt für erhebliche Zeit- und Kostenersparnis, da nur 50% der Bilder erstellt werden.
Nach dem Sequenzier-Prozess erfolgt die bioinformatische Auswertung der detektierten Sequenzen (sog. Reads) indem zuerst die einzelnen Reads gegen das Referenzgenom wie z.B. hg19 aligniert und anschließend in einer Konsensus-Sequenz zusammengefasst werden, um Veränderungen in den sequenzierten DNA-Fragmenten zu identifizieren (Abbildung 1D). Abschließend werden die Sequenzenveränderungen hinsichtlich der klinischen Relevanz beurteilt und die Ergebnisse in einem medizinischen Befund zusammengetragen.

Abbildung 2: 4- und 2-Kanal-SBS-Technologie von Illumina

Autorin: Dr. rer.nat. Ulrike Paul